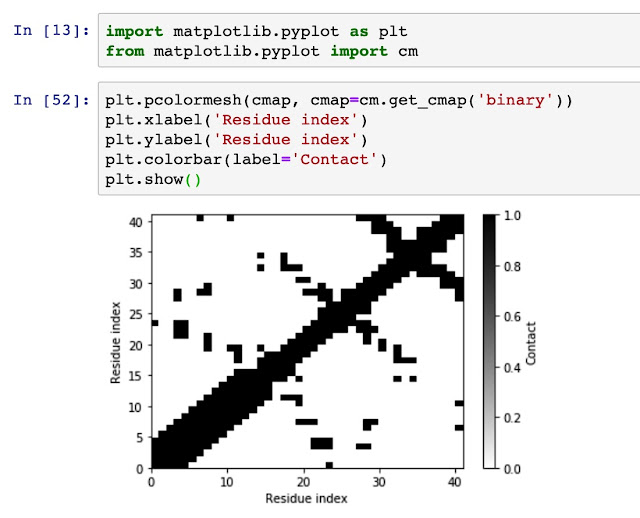
Generate residue-residue contact matrix for MD simulation trajectories Using residue-residue contact map to understand the distance and spatial relationship is important for protein dynamics and protein 3D conformation modeling. Here in this tutorial, I use mdtraj to demonstrate how to generate a contact map from a static single-frame PDB file and visualise it with matplotlib. Taken mini protein gHEEE_02 as an example, we first load the pdb file (generated from rosetta modeling). You could virtually take any protein containing PDB file as an example, such as 5W9F. # load mdtraj package import mdtraj as mt # load the pdb file and transform it into a mdtraj.Trajectory object p = mt.load_pdb("5W9F.pdb") # now we could check some properties of the trajectory object p <mdtraj.Trajectory with 1 frames, 335 atoms, 41 residues, without unitcells at 0x820a38208> Therefore, we know that this protein contains 1 frame, 41 residues....
